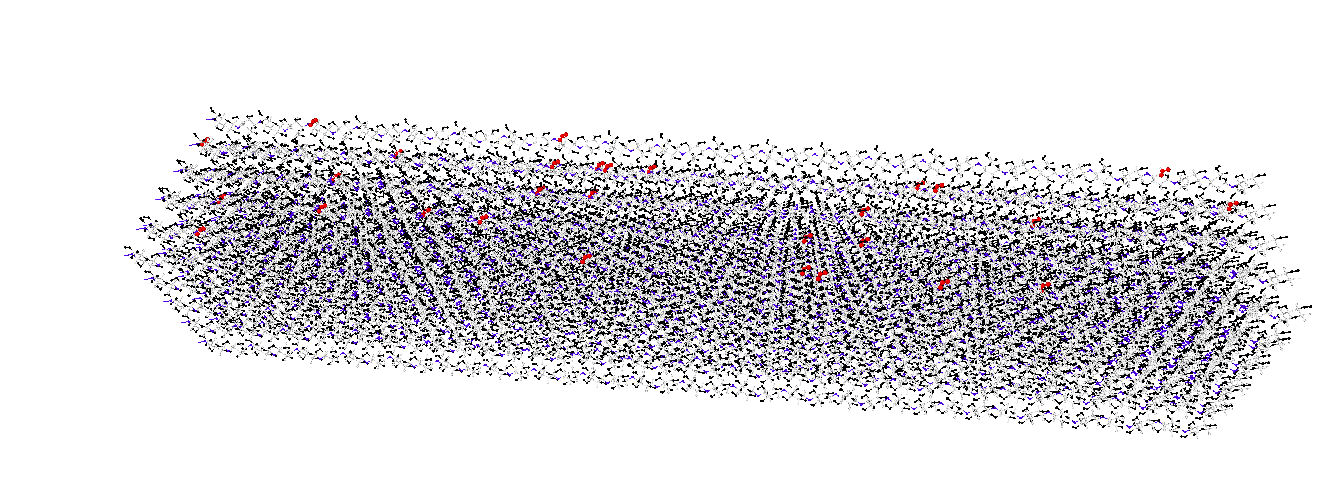
Modeling Oxidized Cellulose Crystals
Published:
Finally figured out the modeling of oxidized cellulose crystals with the CHARMM force field. The approach is similar for other force fields. LAMMPS’s built-in modeling capabilities are really powerful. Mark this down — it deserves a thorough write-up!
Overview
The most common oxidized form of cellulose crystals is TEMPO-oxidized cellulose. Specifically, the hydroxymethyl groups on the cellulose crystal surface are oxidized to carboxyl groups, with an oxidation degree of 0.1~0.5 (note: the degree of sulfonation is generally 0.1~0.2). For the CHARMM force field, some researchers have already achieved cellulose crystal modeling (cellulose-toolkit), but due to the complexity of model construction, studies on surface oxidation are relatively limited. (Existing research has used the OPLS-AA force field: Biomacromolecules 2020, 21, 3069−3080)
Basic Approach
Generally, force field parameters are included inside the data file for polymer systems.\ Two reasons: 1. Polymer systems have many parameters, making it inconvenient to write them all in the input file. 2. The pair_coeff between different atom types is calculated according to the mixing rule (pair_modify), so pair_coeff values differ under different mixing rules. It is simpler to directly write the pair_coeff for individual atom types in the data file and declare the mixing rule in the input file. Alternatively, force field parameters can be included directly using the include command, allowing force field parameters and topology to be read separately. Two notes: 1. Although force field parameters and topology are read separately, the topology must be consistent with the corresponding force field — for example, you cannot read CHARMM force field parameters while using an OPLS-AA topology, because the atom types differ between force fields. 2. Force field parameters can have redundancy! This is important — having extra unused parameters is not a problem. With these two points in mind, modeling oxidized cellulose crystals essentially consists of three parts: + 1 Obtain the Coeff parameter file for oxidized cellulose crystals + 2 Modify the topological structure of the cellulose crystal
The first step can be done by adding force field parameters to the cellulose crystal data file, then using LAMMPS’s built-in write_coeff command to obtain the desired parameter file. (Making good use of LAMMPS’s own modeling capabilities can solve many problems.) Adding force field parameters can be done via a Python script. Note that the input data file must be a standard LAMMPS output data file. python # -*- codeing = utf-8 -*- # @Time :2022/9/9 23:34 # @Author : 得意喵 ~ # @File : addcoeff.py.py # @Software: PyCharm import pandas as pd import numpy as np import re #define functions def findcoeff(data,coeff): for i in range(len(data)): if re.match(coeff,data[i]): return i def coeffformat(n,coeff0): return str(n)+' '+coeff0+'\n' def getcoeffindex(coeff0): s=coeff0.split() return int(s[0]) def addcoeffs(data,n,coeff,n0): for i in range(len(coeff)): data.insert(n+i,coeffformat(n0+i+1,coeff[i])) return data #open files f=open('try.data','r') data=f.readlines() if data[10]=='\n': data.insert(10,'0 impropers\n') data.insert(11,'0 improper types\n') data.insert(findcoeff(data,'Atom')-1,'\n') data.insert(findcoeff(data,'Atom')-1,'Improper Coeffs\n\n') #types and coeffs new_atomtypes=2 new_bondtypes=2 new_angletypes=5 new_dihedraltypes=2 new_impropertypes=1 new_types=[2,2,5,2,1] newmasses=['12.011','15.999'] newpaircoeffs=['0.07000000000000 3.56359487256136 0.07000000000000 3.56359487256136','0.12000000000000 3.02905564167715 0.12000000000000 3.02905564167715'] newbondcoeffs=['525.000 1.2600','200.000 1.4800'] newanglecoeffs=['40.00000 114.00000 50.00000 2.38800','100.00000 132.00000 70.00000 2.22500','50.00000 109.50000 0.00000 0.00000','45.00000 103.00000 0.00000 0.00000','52.00000 108.00000 0.00000 0.00000'] newdihedralcoeffs=['0.0500 6 180 1.00','0.6400 2 180 1.00'] newimpropercoeffs=['96.000 0.00'] new_coeff=[newmasses,newpaircoeffs,newbondcoeffs,newanglecoeffs,newdihedralcoeffs,newimpropercoeffs] coeffindex=[]; index=['Pair Coeffs','Bond Coeffs','Angle Coeffs','Dihedral Coeffs','Improper Coeffs','Atoms'] #change types types=data[3:13:2] for i in range(5): s=types[i].split() s[0]=str(int(s[0])+new_types[i]) types[i]=' '.join(s)+'\n' data[3:13:2]=types #add coeffs for i in range(6): coeffindex.append(findcoeff(data,index[i])) if re.match('\d.',data[coeffindex[i]-2]): data=addcoeffs(data,coeffindex[i]-1,new_coeff[i],getcoeffindex(data[coeffindex[i]-2])) else: data.insert(coeffindex[i]-1,coeffformat(1,new_coeff[i][0])) data1 = open("data_new.data", "w") data1.write("".join(data)) data1.close() f.close() The second step can be implemented using LAMMPS’s modeling capabilities. First, note that the models of cellulose and oxidized cellulose are shown below: 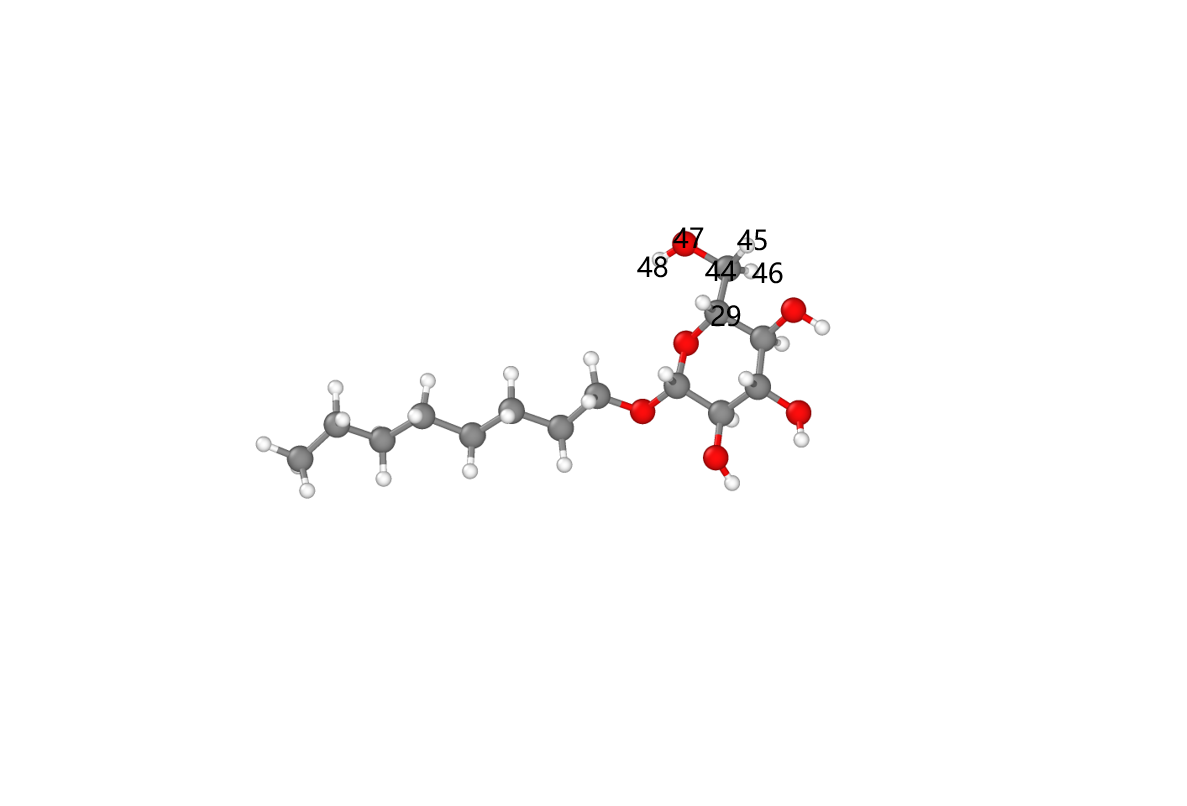
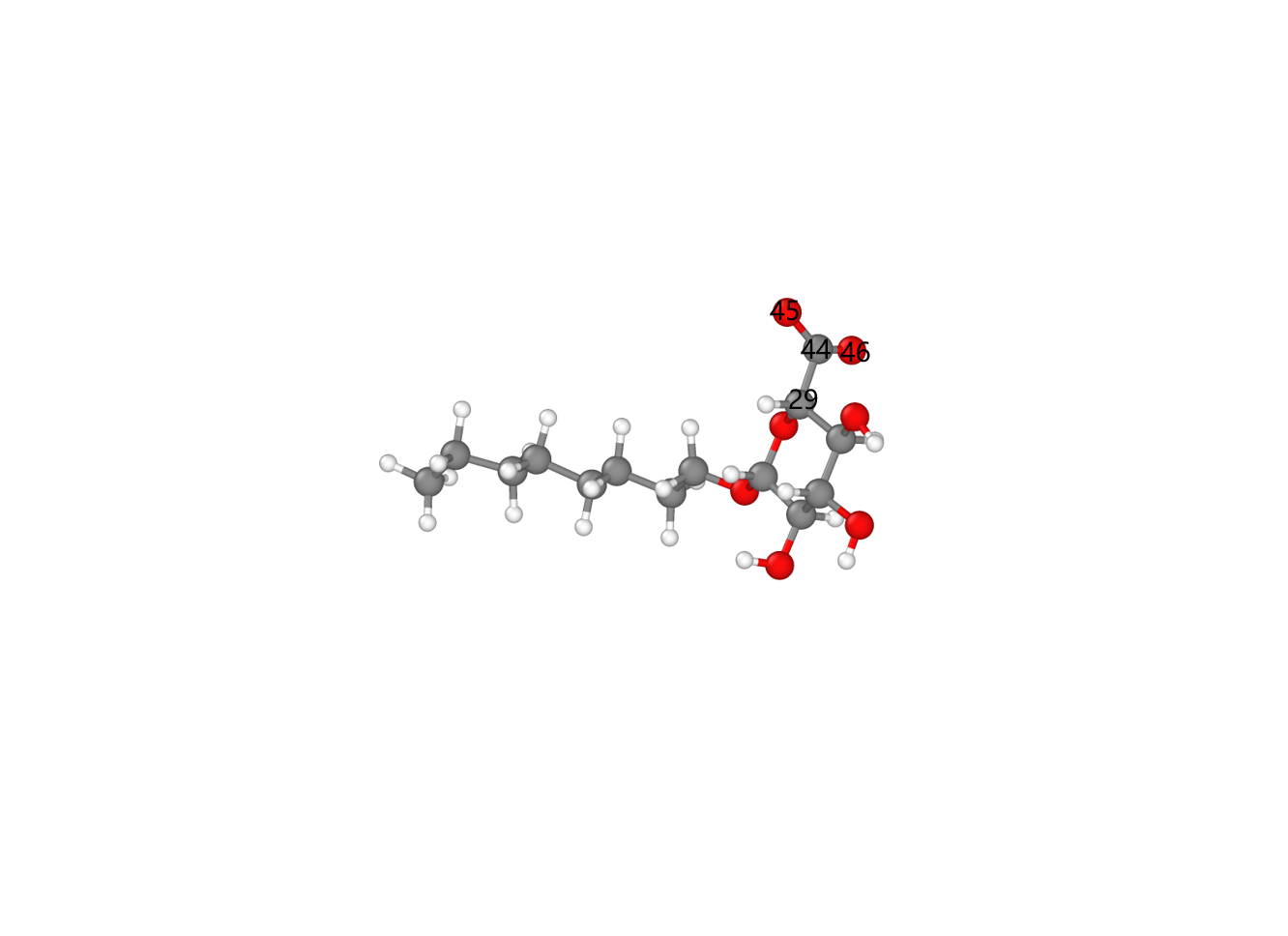 It can be seen that the oxidized form differs from the original only by the absence of the hydroxyl hydrogen atom on the hydroxymethyl group. Interestingly, the two oxygen atoms on the carboxyl carbon after oxidation share the same type, which coincidentally mirrors the two identical hydrogen atoms on the unoxidized hydroxymethyl carbon. Therefore, cellulose oxidation can be achieved simply by deleting the hydroxyl hydrogen and updating the topology information after oxidation (without adding other atoms or redundant topology information, except for adding one out-of-plane dihedral). The topology information before and after oxidation is summarized as follows: Comparison of force field parameters before and after oxidation The different force field parameters were obtained by comparing the polysaccharide data files before and after oxidation generated by CHARMM-GUI. Using LAMMPS’s built-in commands to modify the topological structure of the original data file and assign force field parameters, the CHARMM force field model of the sugar unit after oxidation of hydroxymethyl to carboxyl can be achieved. ```bash units real atom_style full dimension 3 boundary f f f #Force fields bond_style harmonic angle_style charmm dihedral_style charmmfsw improper_style harmonic pair_style lj/charmmfsw/coul/charmmfsh 10.0 12.0 pair_modify mix arithmetic read_data withoutcoeff.data extra/improper/per/atom 5 special_bonds charmm neighbor 2.0 bin neigh_modify every 100 delay 0 check yes include Oxide.coeff # define groups group HO id 48 47 group OO id 45 46 group C id 44 #add coeff # the coeff must be added to data file not the coeff file # it has been done with python code #1 delete_atoms
It can be seen that the oxidized form differs from the original only by the absence of the hydroxyl hydrogen atom on the hydroxymethyl group. Interestingly, the two oxygen atoms on the carboxyl carbon after oxidation share the same type, which coincidentally mirrors the two identical hydrogen atoms on the unoxidized hydroxymethyl carbon. Therefore, cellulose oxidation can be achieved simply by deleting the hydroxyl hydrogen and updating the topology information after oxidation (without adding other atoms or redundant topology information, except for adding one out-of-plane dihedral). The topology information before and after oxidation is summarized as follows: Comparison of force field parameters before and after oxidation The different force field parameters were obtained by comparing the polysaccharide data files before and after oxidation generated by CHARMM-GUI. Using LAMMPS’s built-in commands to modify the topological structure of the original data file and assign force field parameters, the CHARMM force field model of the sugar unit after oxidation of hydroxymethyl to carboxyl can be achieved. ```bash units real atom_style full dimension 3 boundary f f f #Force fields bond_style harmonic angle_style charmm dihedral_style charmmfsw improper_style harmonic pair_style lj/charmmfsw/coul/charmmfsh 10.0 12.0 pair_modify mix arithmetic read_data withoutcoeff.data extra/improper/per/atom 5 special_bonds charmm neighbor 2.0 bin neigh_modify every 100 delay 0 check yes include Oxide.coeff # define groups group HO id 48 47 group OO id 45 46 group C id 44 #add coeff # the coeff must be added to data file not the coeff file # it has been done with python code #1 delete_atoms
delete_atoms group HO bond yes #2 set charge set group OO charge -0.760 set group C charge 0.520 # # # #3 set pair set group OO type 16 set group C type 15 #4 set bond angle dihedral
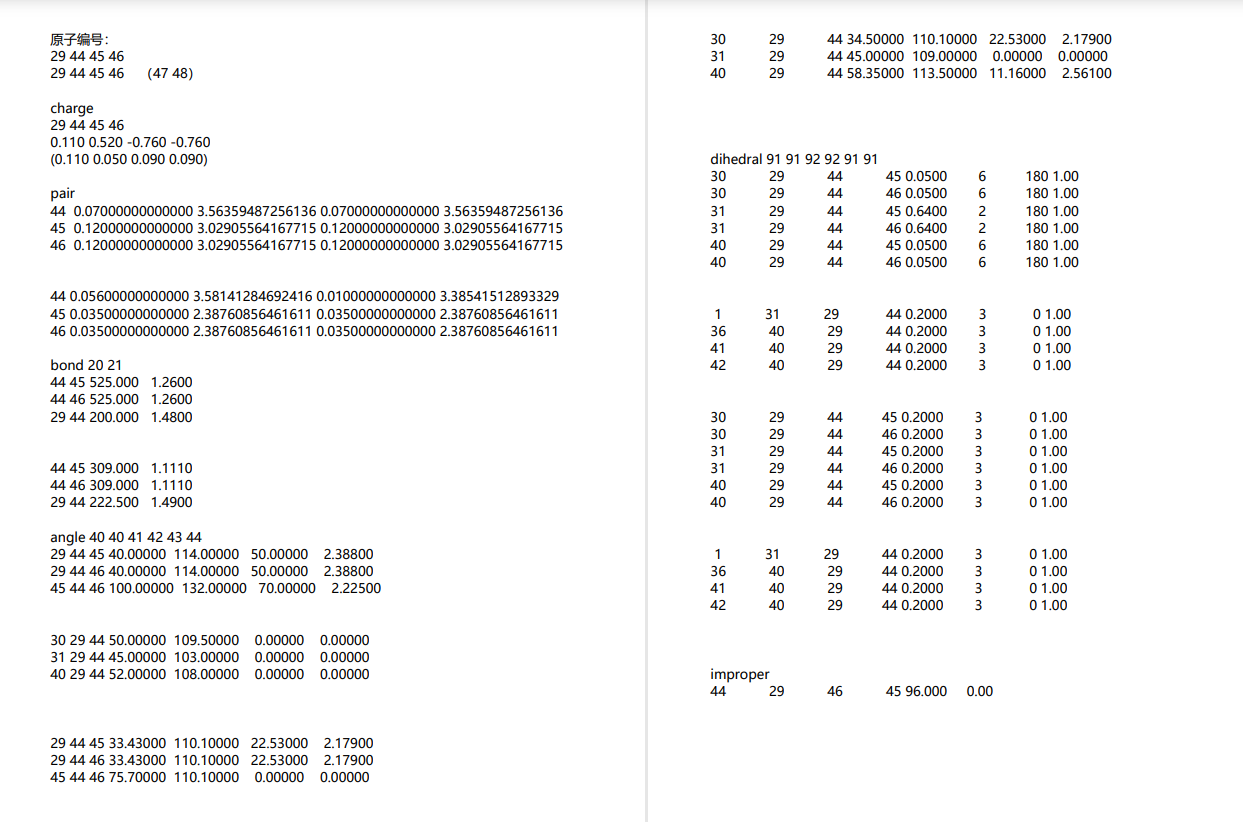{kind=link}
##### Bond
group bondOC id 44 45 46 set group bondOC bond 20 group bondCC id 44 29 set group bondCC bond 21 ########### Angle ########### group angle1 id 29 44 45 set group angle1 angle 40 group angle2 id 29 44 46 set group angle2 angle 40 group angle3 id 45 44 46 set group angle3 angle 41 group angle4 id 30 29 44 set group angle4 angle 42 group angle5 id 31 29 44 set group angle5 angle 43 group angle6 id 40 29 44 set group angle6 angle 44 ########### Dihedral ########### group dihedral1 id 30 29 44 45 set group dihedral1 dihedral 91 group dihedral2 id 30 29 44 46 set group dihedral2 dihedral 91 group dihedral3 id 31 29 44 45 set group dihedral3 dihedral 92 group dihedral4 id 31 29 44 46 set group dihedral4 dihedral 92 group dihedral5 id 40 29 44 45 set group dihedral5 dihedral 91 group dihedral6 id 40 29 44 46 set group dihedral6 dihedral 91 #create and set improper create_bonds single/improper 1 44 29 46 45 write_coeff Oxide.coeff write_data withoutcoeff.data nocoeff ``` At this point, the oxidation from hydroxymethyl to carboxyl is complete. The rest is essentially done. To achieve oxidation of the entire cellulose crystal, simply identify the atom indices of the hydroxymethyl carbons in OVITO, write the indices of the atoms that undergo changes as expressions of the selected carbon indices, and randomly select them for oxidation. (A single loop in LAMMPS can handle this.)
Here is an illustration for a feel of it, haha. The principle is the same. Important notes during oxidation:
1 The random number in a variable changes with each call, so create a new variable to store it!!
2 Delete groups promptly!!
```bash units real atom_style full dimension 3 boundary f f f #Force fields bond_style harmonic angle_style charmm dihedral_style charmmfsw improper_style harmonic pair_style lj/charmmfsw/coul/charmmfsh 10.0 12.0 pair_modify mix arithmetic read_data crystal_nocoeff.data extra/improper/per/atom 500 special_bonds charmm neighbor 2.0 bin neigh_modify every 100 delay 0 check yes # write_data crystal_new.data include crystal.coeff
variable NS loop 30
label loop variable seed equal ${NS}^2+2${NS}+3932 variable a290 equal round(random(0,1,${seed})180)*42+34443 variable a29 equal ${a290} print “ ${a29} “ # variable a29 equal 36921 variable a44 equal ${a29}+14 print “ ${a29} “ variable a45 equal ${a29}+15 print “ ${a29} “ variable a46 equal ${a29}+16 variable a47 equal ${a29}+17 variable a48 equal ${a29}+18
define groups
group HO id ${a47} ${a48} group OO id ${a45} ${a46} group C id ${a44} #add coeff # the coeff must be added to data file not the coeff file # it has been done with python code #1 delete_atoms
delete_atoms group HO bond yes #2 set charge set group OO charge -0.760 set group C charge 0.520 # # # #3 set pair set group OO type 12 set group C type 11 #4 set bond angle dihedral
##### Bond
group bondOC id ${a44} ${a45} ${a46} set group bondOC bond 16 group bondCC id ${a44} ${a29} set group bondCC bond 17
##### Angle
group angle1 id ${a29} ${a44} ${a45} set group angle1 angle 33 group angle2 id ${a29} ${a44} ${a46} set group angle2 angle 33 group angle3 id ${a45} ${a44} ${a46} set group angle3 angle 34 group angle4 id 30 ${a29} ${a44} set group angle4 angle 35 group angle5 id 31 ${a29} ${a44} set group angle5 angle 36 group angle6 id 40 ${a29} ${a44} set group angle6 angle 37 ########### Dihedral ########### group dihedral1 id 30 ${a29} ${a44} ${a45} set group dihedral1 dihedral 84 group dihedral2 id 30 ${a29} ${a44} ${a46} set group dihedral2 dihedral 84 group dihedral3 id 31 ${a29} ${a44} ${a45} set group dihedral3 dihedral 85 group dihedral4 id 31 ${a29} ${a44} ${a46} set group dihedral4 dihedral 85 group dihedral5 id 40 ${a29} ${a44} ${a45} set group dihedral5 dihedral 84 group dihedral6 id 40 ${a29} ${a44} ${a46} set group dihedral6 dihedral 84 #create and set improper create_bonds single/improper 1 ${a44} ${a29} ${a46} ${a45}
group HO delete group OO delete group C delete group bondOC delete group bondCC delete group angle1 delete group angle2 delete group angle3 delete group angle4 delete group angle5 delete group angle6 delete group dihedral1 delete group dihedral2 delete group dihedral3 delete group dihedral4 delete group dihedral5 delete group dihedral6 delete # delete_atoms group HO bond yes next NS jump SELF loop write_data Oxide.data nocoeff ```