

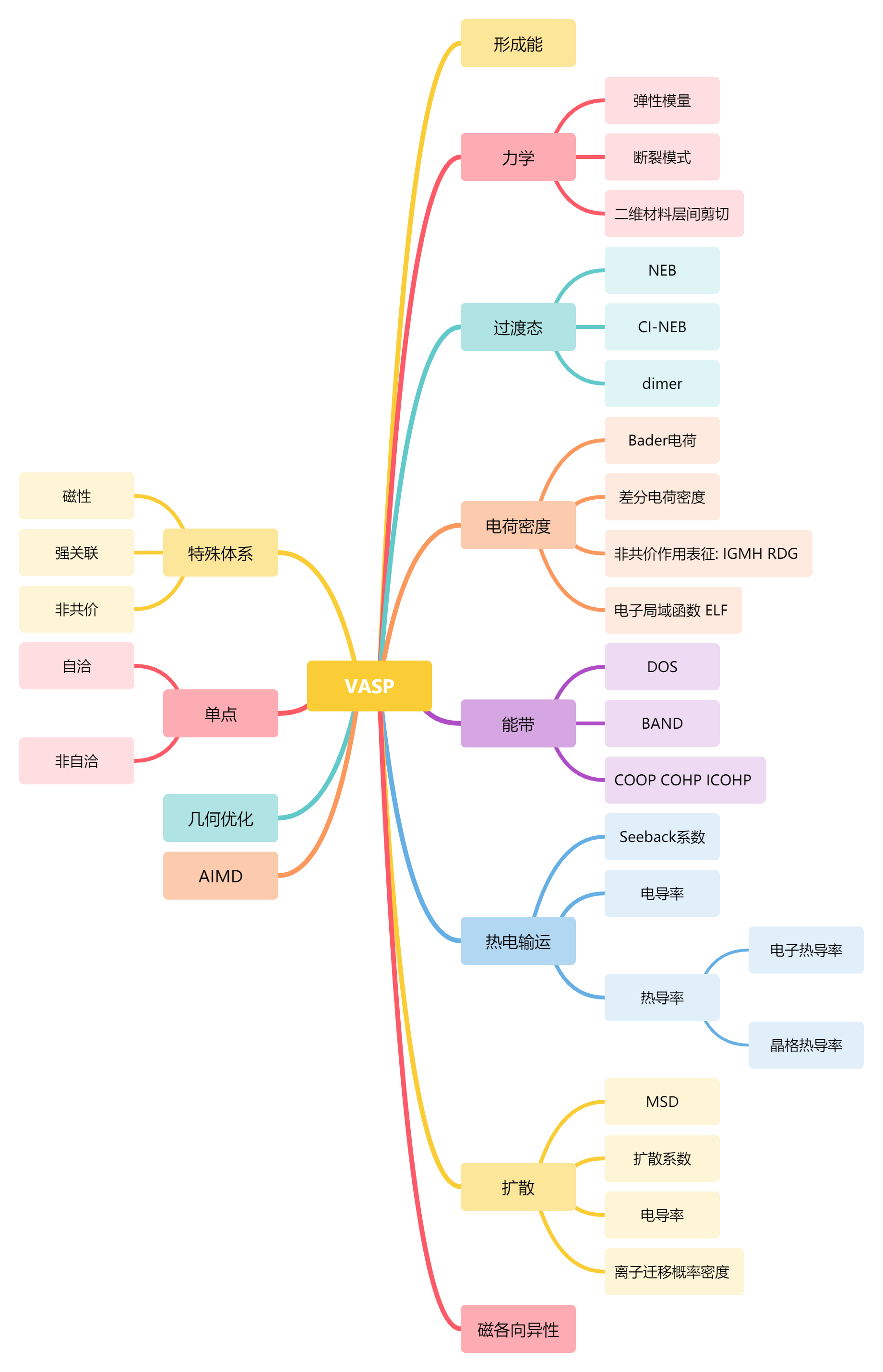
VASP计算简单分为如下几类:
Global
IO parameters
- ISTART = 1 read wavefunction
- ICHARG = 1 read charge density (11 for DOS and BAND)
- LWAVE = .TRUE. write wavefunction
- LCHARG = .TRUE. write charge density
- LELF = .FALSE. write ELFCAR
Precision parameters
- ENCUT = 1.3*ENMAX in POTCAR
- #NELECT = ? number of valence electrons (Background charge)
- ADDGRID = .TRUE. oscillations the charge density
- PREC = Accurate
- LREAL = Auto projection done in real space
Parallel parameter
- KPAR = number of cores/KPAR for one k-point
- NCORE = cores/KPAR/NPAR
-#NPAR =
SP
- ISMEAR = 0/1/-5
- SIGMA = 0.02~0.05/0.2/\
- NELM = 200
- EDIFF = 1e-5~1e-4
- # NEDOS = 2000
- # LORBIT = 11
OPT
- NSW = 2000
- IBRION = 2(CG) 0(MD) 3(CI-NEB)
- ISIF = 2 (ions) 3 (ions/shape)
- EDIFFG = -1e-2 ~ -5e-2 force smaller 0.02A/eV
- ISYM = 2 use symmetry 0 for AIMD
- ALGO = VeryFast (Normal for magnetic system)
MAGMOM
- ISPIN = 2 open spin 1 close spin
- MAGMOM = n1*uB1 n2*uB2 n1*uB1 n2*uB2 n3*uB3 n4*uB4
- VOSKOWN = 0 (1 for PW91 function)
- LASPH = .TRUE. (Non-spherical elements, d/f convergence)
- GGA_COMPAT = .FALSE. (Apply spherical cutoff on gradient field FALSE for magnetic anisotropy)
- AMIX = 0.2 (Mixing parameter to control SCF convergence)
- BMIX = 0.0001 (Mixing parameter to control SCF convergence)
- AMIX_MAG = 0.4 (Mixing parameter to control SCF convergence)
- BMIX_MAG = 0.0001 (Mixing parameter to control SCF convergence)
DFT+U
- LDAU = .TRUE. (Activate DFT+U)
- LMAXMIX = 4 (For d elements increase LMAXMIX to 4, f: LMAXMIX = 6)
- LDAUTYPE = 2 (Dudarev, only U-J matters)
- LDAUL = d/f d/f d/f d/f (Orbitals for each species 1p 2d 3f -1none)
- LDAUU = U1 U2 U3 U4
- LDAUJ = 0 0 0 0
NEB
- ICHAIN = 0 (Open NEB)
- LCLIMB = .TRUE. (Choose CI-NEB)
- IOPT = 1/2/7 (7:Fast Intertial Relaxation Engine; 2:CG)
- POTIM = 0 (Use method provided by CI-NEB)
- IMAGES = 8 (Numbers of interpolation points)
- SPRING = -5 (Spring force)
不同体系需要设置的参数
- 体系性质:SIGMA; ISMEAR; DFT+U; MAGMOM
- 收敛参数:EDIFF; EDIFFG; NSW; NELM
- 非自洽计算: ICHARG; NEDOS; LORBIT
- 并行参数:NCORE; KPAR
自由能矫正
NFREE = 2
IBRION = 0.015
自动化处理脚本
- 优化+单点
- 单点+非自洽 能带
- 自由能矫正
经验总结-liujc
我个人通常习惯做分步结构优化。在第一步结构优化中,使用较低精度,同时设置 ISMEAR = 0 + SIGMA = 0.1,查看结构优化最后在 OUTCAR 中给出的 EENTRO 值。然后通过 EENTRO 值除以体系原子数去判断体系是半导体和金属:
( EENTRO / 原子数 ) 大于 1meV,体系为金属。
( EENTRO / 原子数 ) 小于 0.1meV,体系为半导体。这个值非常小时,比如 0.000001,体系带隙一般较大。
( EENTRO / 原子数 ) 介于 1meV 和 0.1meV,目前还没有具体测试过,不确定。
这一判断的依据是:对于金属体系,ISMAER = 0 会使用 Gaussian smearing,导致在费米面处出现分数占据,进而导致 EENTRO 值不为 0 。而对于较大带隙的半导体,SIGMA = 0.1 还无法导致费米面处出现分数占据,EENTRO 值保持为 0(若增大 SIGMA 到一个很大的值时,EENTRO 也将不再保持为 0,但此时的计算会存在问题)。
这一判断方法是我个人的经验,目前在使用过程中大部分情况下都能给出正确的结果,但不保证对所有体系所有情况都可用。比如在弛豫过程中晶格变化非常大时,用于自洽计算对应的平面波截断球已经严重变形,可能会导致离子步的自洽收敛到的状态本身就有很大的误差。此时再在有误差的自洽结果上用这一方法进行判断是否可行,我不确定。不过解决办法也简单,再用弛豫的结构做一次弛豫就可以使用这一方法进行判断了。
本文采用CC-BY-SA-3.0协议,转载请注明出处
作者: 得意喵~
作者: 得意喵~